 Concept Figure
Concept Figure
Abstract
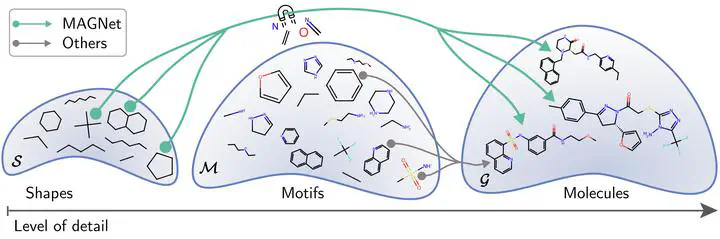
Recent advances in machine learning for molecules exhibit great potential for facilitating drug discovery from in silico predictions. Most models for molecule generation rely on the decomposition of molecules into frequently occurring substructures (motifs), from which they generate novel compounds. While motif representations greatly aid in learning molecular distributions, such methods struggle to represent substructures beyond their known motif set. To alleviate this issue and increase flexibility across datasets, we propose MAGNet, a graph-based model that generates abstract shapes before allocating atom and bond types. To this end, we introduce a novel factorisation of the molecules’ data distribution that accounts for the molecules’ global context and facilitates learning adequate assignments of atoms and bonds onto shapes. Despite the added complexity of shape abstractions, MAGNet outperforms most other graph-based approaches on standard benchmarks. Importantly, we demonstrate that MAGNet’s improved expressivity leads to molecules with more topologically distinct structures and, at the same time, diverse atom and bond assignments.
Leon Hetzel
PhD Student in Machine Learning and Computational Biology
My research interests include generative modelling, single-cell genomics, and drug discovery.